Structure Conversion
An important feature of HQS Molecules is conversion of two-dimensional molecular structural formulas into three-dimensional geometries, as exemplified below for the glucose molecule. Functionality of RDKit and Open Babel is built upon in order to obtain results more reliably than with either software on its own. Generated three-dimensional structures are verified against the input, increasing trustworthiness for automated workflows.
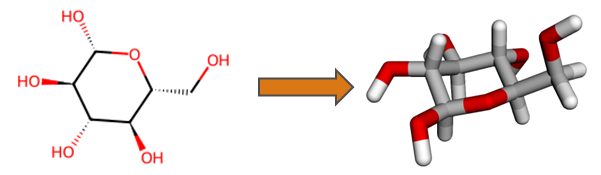
Simple 2D to 3D Conversion
Input: SMILES strings and Molfiles are supported as input for the functions smiles_to_molecule and molfile_to_molecule, respectively.
Output: Both functions return a Molecule object containing three-dimensional atomic coordinates. In addition, the returned object contains the overall molecular charge, which is always included in a Molfile or a SMILES string. The spin multiplicity field is set to None, as it cannot be derived unambiguously from the input.
The structure conversion employs the respective feature of RDKit as its first choice, and uses Open Babel as a backup in case that structure conversion with RDKit fails. After generating a three-dimensional structure, a bonding graph is determined using distance criteria and used to verify the generated structure against the input. If the composition and the bonding graphs do not match, then the structure is rejected.
Having the SMILES string of a molecule at hand, it is straightforward to create its 3D representation with a function call. For example, we may first obtain the SMILES string of a molecule from PubChem:
from hqs_molecules import PubChem, smiles_to_molecule
pc = PubChem.from_name("propane")
print(pc.smiles)
# CCC
The three-dimensional structure is created from the string with a call to smiles_to_molecule:
mol = smiles_to_molecule("CCC")
Conversion of a structural formula stored in a Molfile ("my_molecule.mol" in this example) proceeds similarly:
from hqs_molecules import molfile_to_molecule
mol = molfile_to_molecule("my_molecule.mol")
An optional check can be carried out with either of the conversion functions by supplying a molecular formula as an argument. The conversion fails if the input does not match the provided formula. In that case, the formula needs to be represented as a MolecularFormula object.
from hqs_molecules import MolecularFormula, PubChem, smiles_to_molecule
pc = PubChem.from_name("propane")
# succeeds
mol = smiles_to_molecule(pc.smiles, formula=pc.formula)
# raises an exception
mol = smiles_to_molecule(pc.smiles, formula=MolecularFormula.from_str("C3H7-"))
The code above will raise an error in the last line since a wrong formula of propane is provided.
Utilities for RDKit
The HQS Molecules module includes convenience functions to create RDKit Mol objects from SMILES strings, Molfiles, or XYZ files. These objects represent molecular information within the RDKit package.
Both the smiles_to_rdkit and molfile_to_rdkit functions accept an argument addHs. By default, it is set to True, causing explicit hydrogen atoms to be added in the generated object. Setting addHs = False suppresses the addition of explicit hydrogens; only hydrogens that were already explicitly represented within a Molfile are retained.
from hqs_molecules import smiles_to_rdkit
# The object generated contains 11 atoms.
rdkit_mol = smiles_to_rdkit("CCC")
# The object generated contains 3 atoms.
rdkit_mol = smiles_to_rdkit("CCC", addHs=False)
When creating an RDKit Mol object from an XYZ file, this option does not apply as the hydrogen atoms always have to be represented explicitly. However, the xyzfile_to_rdkit function accepts another important argument (apart from the XYZ file name or path) which is the charge. This argument is necessary to specify the total net charge of the molecule in order to find the correct atomic connectivity and it is set to 0 by default for convenience. Since an XYZ file does not contain any information about chemical bonds by itself, this evaluation is done inside the xyzfile_to_rdkit function using RDKit.
For the following example, assume we have two valid XYZ files called benzene.xyz with the three-dimensional structure of a benzene molecule and nh4.xyz with the structure of an ammonium cation.
from hqs_molecules import xyzfile_to_rdkit
# The object generated contains 12 atoms, 6 single bonds, and 6 aromatic bonds.
rdkit_mol = xyzfile_to_rdkit("benzene.xyz")
# This will raise a `ValueError`
rdkit_mol = xyzfile_to_rdkit("nh4.xyz")
# This works and the object generated contains 5 atoms and 4 single bonds.
rdkit_mol = xyzfile_to_rdkit("nh4.xyz", charge=1)
Expert Usage
Features described in the remainder of this section are only intended for expert usage.
An RDKit Mol object can be converted to a three-dimensional structure by passing it to the function rdkit_to_molecule. It is a low-level function that calls RDKit without resorting to Open Babel as a backup. Nonetheless, it performs a consistency check for the generated structure. If the RDKit Mol object was created without the addition of explicit hydrogens (addHs = False), this conversion may fail due to a composition mismatch.
The low-level functions to perform structure conversion using only Open Babel are available via smiles_to_molecule_obabel and molfile_to_molecule_obabel. These functions require a SMILES string or a Molfile as their input, respectively. A separate consistency check of the generated structure is also performed here.